Our lab's research centers on understanding the pathological effects of damaging extrinsic stimuli on mucosal epithelia. Her laboratory is working to understand the pathogenesis ocular surface damage in patients with autoimmune disease, as well as the molecular mechanisms that promote the transformation of airway mucosal epithelial cells to tumor cells in response to cigarette smoke. Although seemingly different, these studies share in common the need for a better understanding of the molecular events that contribute to pathological alteration of mucosal epithelia, and their potentially devastating consequences. Our research program involves both clinically based, human studies to characterize key components of mucosal defense, as well as studies to decipher the mechanisms whereby they modulate disease.
Molecular Mechanisms of Squamous Metaplasia in Keratinizing Ocular Surface Disease
Pathological keratinization of the ocular surface, also known as squamous metaplasia (SQM) causes a debilitating and vision-threatening disease that can eventually progress to complete corneal opacification and blindness
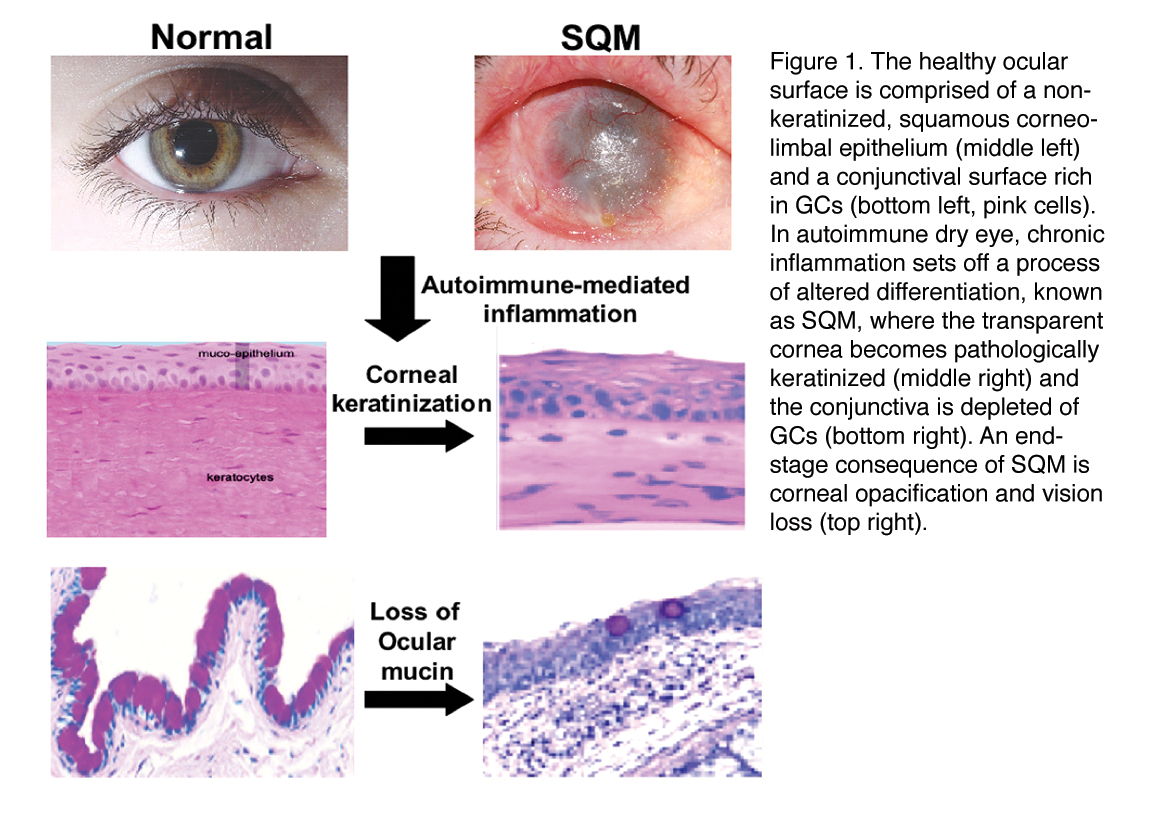
It occurs in severe, autoimmune-mediated disorders, such as Stevens-Johnson syndrome (SJS), ocular cicatricial pemphigoid (OCP) and Sjögren’s syndrome (SS). These diseases present some of the most challenging clinical cases facing eye care today and targeted therapies that can be used as treatment alternatives to systemic immunosuppression are not currently available.
Very little is known about the specific mechanisms mediating SQM in the setting of autoimmunity. Over the past several years, we have worked to identify mediators of chronic inflammation that set off the process of keratinization. They identified small proline rich protein 1B (SPRR1B), a protein specific to keratinized epithelia, as a surrogate biomarker for keratinizing SQM in human patients with SS that was highly correlated with clinical disease severity and inflammation.

Using tear proteomics they identified cytokines upregulated in the tears of patients with active ocular cicatricial pemphigoid and discovered that increased protein levels of interleukin 1 alpha (IL1α) and IL1β were correlated with dry eye symptoms in SS patients.
In an animal model of autoimmune dry eye, ocular SQM was dependent on autoreactive CD4+ T cell infiltration and local signaling via the IL-1 receptor.2 Macrophages are a major source of IL-1 during inflammation and we identified a pathological role for macrophages in autoimmune dry eye. They depleted macrophages using clodronate liposomes both locally, via subconjunctival injection, and systemically, via intraperitoneal injection. In mice with autoimmune dry eye, subconjunctival clodronate restored ocular surface integrity while systemic treatment normalized tear secretion. These studies suggested that depletion of macrophages in the setting of autoimmune dry eye significantly reduced inflammation and ocular damage while increasing tear secretion
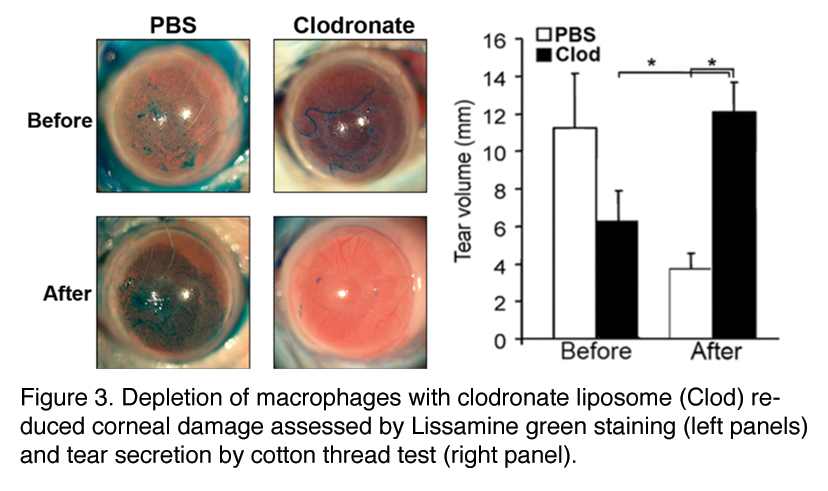
In future studies, we will explore the mechanism whereby macrophages contribute to autoimmune-mediated ocular surface disease and the usefulness of modifying macrophage function or blocking their recruitment as a novel treatment strategy.
Most recently, our group identified a role for stem cells in SQM pathogenesis. They discovered that corneal stem cells become activated in SQM disease and this activation drives the aberrant program of cellular differentiation that leads to SQM. Altered stem cell activation appears to occur when corneal cells lose their master regulator, Pax6. They are testing the hypothesis that ocular stem cells and Pax6 are the cellular and molecular links between chronic inflammation and ocular SQM disease. In preliminary studies, forced expression of Pax6 in diseased eyes using adenoviral transfer normalized stem cell activity and restored the tissue phenotype. The biological significance and therapeutic implications of this work are evident and strategies geared to restore ocular health at the stem cell level may prove helpful in the treatment of autoimmune diseases that promote ocular surface disease.
Sjögren’s International Collaborative Clinical Alliance (SICCA)
Our experience in the area of autoimmune ocular surface disease extends into the clinical arena through our work with SICCA. As a SICCA investigator since 2006, Dr. McNamara collects clinical data and biospecimens for the Sjögren’s Syndrome Registry and participates in the preparation of scientific manuscripts. Most recently, the SICCA group established important associations of the labial salivary gland biopsy focus score with phenotypic ocular and serological components of SS,4 as well as developing new classification criteria for the diagnosis of SS.
Smoke-Induced Lung Cancer: Role of EGFR, MUC1 and Catenins
Smoking represents the single most important carcinogenic exposure and is the leading cause of cancer-related mortalities. An estimated 219,440 Americans were diagnosed with lung cancer in 2009, 87% of which were directly attributable to cigarette smoke. In order to identify drug targets that can be exploited by pharmaceutical companies to alleviate and/or prevent lung cancer caused by tobacco smoke, our lab uses a novel, 3-dimensional experimental model of stratified bronchial epithelial cells to simulate the in vivo airway. Cells are exposed to smoke using a fast, easy, and reproducible method, which offers the opportunity to perform manipulations at a molecular level. Using this model, they have identified an essential role for structural units that maintain the polarity of airway epithelial cells in the pathogenesis of lung cancer. Membrane-bound mucins on the apical cell surface and sub-apical junctional complex components E-cadherin, β-catenin and p120 catenin, establish a structural barrier that protects the airway from infectious, inflammatory and noxious stimuli
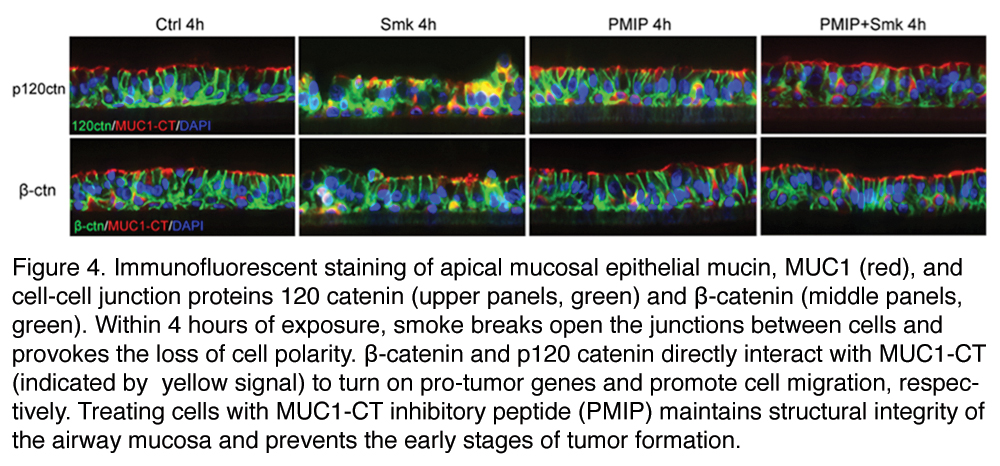
As an early step in tumorigenesis, tobacco smoke disrupts cell-cell interactions, liberating β-catenin and p120 catenin from their junctional complexes. Once liberated, catenins function as tumor promoting oncogenes through their interactions with the cytoplasmic tail of MUC1 (MUC1-CT). MUC1-CT chaperones β-catenins to the nucleus where it turns on pro-tumor genes in response to smoke,6 while 120 catenin promotes migration of airway epithelial cells.7 In addition to its role in cell migration, p120catenin interacts the MUC1-CT to induce the transformation of normal mucosal epithelial cells to tumor cells. Using a MUC1-CT blocking peptide, PMIP, the laboratory was able to restore polarity of the airway mucosa and prevent early events that promote tumor formation.8 A deeper understanding of smoke-induced cell transformation and migration may provide a platform for screening new drug candidates that suppress tumor formation, progression and migration in metastatic disease.
